
(PRAWIE) WSZYSTKO O REAKCJI LANDOLTA
** ostatnia aktualizacja: 4.11.2013 **
Hans Heinrich Landolt (1831-1910), fizykochemik niemiecki poch. szwajcarskiego. Od 1857 profesor uniwersytetu w Bonn, od 1865 w Akwizgranie, od 1880 w Berlinie. Badał związki arsenu, antymonu, czynność optyczną związków organicznych, wykonał pomiary potwierdzające słuszność zasady zachowania masy w reakcjach chemicznych. Opracował obszerne tabele właściwości fizykochemicznych związków organicznych.
Reakcją Landolta nazywane są różne F reakcje "zegarowe" pomiędzy jodanami a niektórymi reduktorami. Najbardziej znana jest reakcja z udziałem wodorosiarczynu, chociaż opisywana była również reakcja jodanów z arseninami. Atrakcyjność reakcji Landolta wynika przede wszystkim z widowiskowego, jeszcze bardziej nietypowego niż u innych reakcji zegarowych, przebiegu czasowego procesów, skomplikowanego jej przebiegu, oraz z niezliczonej wprost możliwości modyfikacji pokazu. Tu zostanie zaproponowanych ponad 20 wersji tej reakcji. Kto pomoże wymyślić dodatkowe?
W odróżnieniu od innych typowych reakcji "zegarowych", reakcję Landolta uruchamia się z użyciem nie trzech, ale zaledwie dwóch substratów (oraz pomocniczego roztworu skrobi, jako wskaźnika).
roztwór jodanu: 15 g KIO3 (lub 15 g NaIO3 x H2O) - w litrze wody (nieco dalej będzie podany przepis bardziej "oszczędny")
roztwór wodorosiarczynu: 15 g NaHSO3 # oraz 3 g skrobi (zagotowanej oddzielnie z małą ilością wrzącej wody) - w litrze wody
już teraz warto przygotować roztwory, które będą przydatne w kolejnych modyfikacjach:
roztwór soli rtęci: 3 g HgCl2 (sublimat, silna trucizna!) ## - w litrze wody
roztwór 0,2 M NaOH
0,2-procentowy roztwór błękitu bromofenolowego
# wodorosiarczyn można zastąpić nieco mniejszą ilością pirosiarczynu Na2S2O5; pirosiarczyn w wodnym roztworze szybko hydrolizuje do wodorosiarczynu. W praktyce jednak (może z powodu obecności zanieczyszczeń w handlowym produkcie) użycie wodorosiarczynu daje znacznie lepsze wyniki.
## związki rtęci są silnie trujące i szkodliwe dla środowiska. Zgodnie z przepisami odpady takie powinny być gromadzone i przerabiane. Proponuję, aby o szczegóły zapytać na najbliższej uczelni prowadzącej studia chemiczne. Interesujące, co powiedzą... A może ich rtęć nie jest toksyczna?
wersja "klasyczna"
W zlewce (pożądane jest mieszadło magnetyczne) zmieszać: 50 ml roztworu jodanu, 350 ml wody, oraz 50 ml roztworu siarczynu (te proporcje odpowiadają nadmiarowi jodanu. Dokładniej opisane będzie to podczas miareczkowych wersji reakcji Landolta). Naczynie należy bardzo uważnie obserwować, gdyż efekt końcowy (powstanie ciemnego, prawie czarnego zabarwienia) następuje szybciej, niż mrugnięcie okiem! Czas reakcji: ok. 2 minuty.
Również demonstrator może dokonywać swoich własnych obserwacji. Trudno zapomnieć widok zapełnionego audytorium, z dziesiątkami oczu wpatrzonych w zlewkę na stole demonstracyjnym! Jeden z autorów książki o pokazach chemicznych, tak to opisuje:
"Jak wiemy, nie ma w sali wykładowej większej ciszy nad tę, która zapada na czas pierwszych chwil chemicznego pokazu. Tych chwil zamilknięcia w spontanicznym, wytężonym skupieniu zazdroszczą nam reżyserzy i dyrektorzy artystyczni filharmonii".


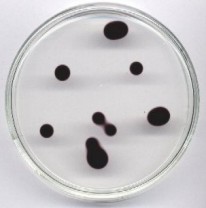
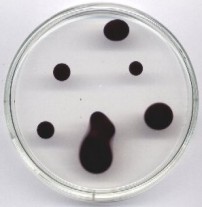
Wprost idealne byłoby tu załączenie animowanego filmu ilustrującego przebieg reakcji. Na razie pozostaje jednak tylko oglądanie poniższych skanów, aby obejrzeć kolejne fazy reakcji. (Przyznam się prywatnie, że poniższe obrazki najbardziej przypominają mi F szwajcarską sygnalizację kolejową).
dlaczego tak bardzo raptownie?
Po zmieszaniu zakwaszonych roztworów: jodanu IO3– (bezbarwny utleniacz) oraz siarczynu SO32– (bezbarwny reduktor), biegnie powolna reakcja redoks, w wyniku której powoli powstają bezbarwne aniony jodkowe I– ### :
IO3– + 3 HSO3– → I– + 3 SO42– + 3 H+ ......(powoli).(1)
### przypominam, że aniony jodkowe I– ze skrobią nie reagują; niebiesko zabarwiony kompleks tworzy ze skrobią wolny pierwiastkowy jod I2. Zupełnie poprawnie: do wytworzenia niebieskiego zabarwienia potrzebne prócz jodu, są jednak również aniony jodkowe. Ostatecznie przyjmuje się, że ze skrobią reagują aniony kompleksu o składzie: I5–.
W trakcie powolnego powstawania anionów jodkowych w roztworze znajdują się ciągle znaczne ilości zarówno jodanu, jak i siarczynu. Powstające powoli aniony jodkowe utleniane są szybko przez jodan - do wolnego jodu:
5 I– + IO3– + 6 H+ → 3 I2 + 3 H2O ......(szybko).(2)
Powstający wolny jod nie zdąży ze skrobią utworzyć granatowego kompleksu, gdyż jest natychmiast redukowany przez istniejący ciągle w roztworze siarczyn:
I2 + HSO3– + H2O → 2 I– + SO42– + 3 H+ ......(bardzo szybko).(3)
procesy te biegną kolejno i równocześnie do chwili zużycia jednego z substratów. Dopóki w roztworze znajduje się niezużyty siarczyn, wszystkie powstające związki jodu przekształcane są w postać zredukowaną (aniony jodkowe). W klasycznej wersji używa się nadmiaru utleniacza. Powstałe aniony jodkowe (w tym momencie już w roztworze nie ma siarczynu) utleniane są przez jodan - do jodu I2 , który ze skrobią tworzy raptownie granatowe zabarwienie.
I2 + skrobia → granatowy kompleks......(dość szybko)
** http://www.youtube.com/watch?v=KWJpKNQfXWo **
Początkowo zarówno jodan jak i siarczyn zużywane są bardzo powoli: jedynie w wyniku powolnej reakcji (1). W miarę trwania procesu, zarówno jodan, jak i siarczyn w coraz większym stopniu zużywane są na drodze pośredniej "via jodki": poprzez szybkie reakcje (2) i (3). Tempo zużywania substratów rośnie. Opisany tu mechanizm jest typowym mechanizmem katalizy: reakcja może zachodzić albo bezpośrednio (powoli), albo inną (szybką) drogą, poprzez związki pośrednie, które zostają po jej zajściu odtworzone. Tę funkcję pełnią w reakcji Landolta aniony jodkowe; ich rolę można porównać do czółenka tkackiego. Należy pamiętać, że aniony jodkowe produkowane są ciągle przez zachodzącą cały czas reakcję (1). Reakcja Landolta jest więc reakcją, której produktem jest substancja będąca jednocześnie jej katalizatorem; taki proces nazywany jest reakcją autokatalityczną. Nieco później wykażemy istnienie jeszcze jednego mechanizmu autokatalizy w tym procesie. Wszystko to powoduje, że reakcja Landolta ma zupełnie nietypowy przebieg: powolna na początku, lawinowo przyspiesza (!) w miarę upływu czasu. W jednej z ostatnich modyfikacji wykażemy w efekciarskim doświadczeniu, że aniony jodkowe są rzeczywiście katalizatorem reakcji Landolta. Dopiero pod koniec tempo reakcji nieco maleje na skutek zużywania substratów.
Lawinowo przyspieszający charakter reakcji Landolta można doskonale zaobserwować uruchamiając którąś z innych typowych reakcji zegarowych: Harcourta (KI + H2O2 + Na2S2O3) lub formalinowej (HCHO + Na2SO3 + NaHSO3) i porównując raptowność efektu końcowego, z reakcją Landolta o podobnym czasie ich zachodzenia. Końcowy efekt w reakcji Landolta przypada na ostatni, silnie rozpędzony jej etap, podczas gdy w typowych reakcjach nie-autokatalitycznych - przypada na bardzo już powolne stadia końcowe.
autokataliza: kinetyczna nieliniowość chemiczna
Reakcja Landolta jest dobrym modelem ilustrującym pojęcie nieliniowości kinetycznej. W tej autokatalitycznej reakcji biorą udział jony jodkowe powstałe w wyniku reakcji (1), konsumowane następnie w reakcji (2) i odtwarzane w reakcji (3). Podsumujmy dwa ostatnie procesy, porównując współczynniki przy anionach jodkowych:
5 I–
+ IO3– + 6 H+ → 3 I2 + 3 H2O..... (2),
tzw. "reakcja Dushmana"
3 I2
+ 3 HSO3– + 3 H2O → 6 I– + 3 SO42–
+ 9 H+..... (3)
5 I– + IO3– + 3 HSO3– → 6 I– + 3 SO42– + 3 H+ , w skrócie:
5 I– → [...] → 6 I–
Aniony jodkowe są więc w autokatalitycznej reakcji Landolta powielane. Współczynnik "pomnażania" wynosi tu 1,2 (120%) na jeden pełen cykl przemian. Stężenie anionów jodkowych ma zasadniczy wpływ na szybkość reakcji Landolta, stężenie jodków znajduje się więc w kinetycznym równaniu procesu (2), istotnego dla przebiegu autokatalizy:
v = k2 x [IO3–] x [I–] 2 x [H+] n ........wartość n zawarta jest pomiędzy 2 a 3.... .. (4)
W tym momencie warto zaznaczyć, że postać algebraiczna doświadczalnego równania kinetycznego, może mieć formę nie mającą związku ze współczynnikami stechiometrycznymi w równaniu sumarycznym reakcji.
Charakterystyczne, że w tym równaniu kinetycznym stężenie autokatalizatora występuje w potędze o wykładniku większym od jedności. Takie równania określane są w matematyce jako równania nieliniowe. Reakcja autokatalityczna jest więc przykładem procesu chemicznego o nieliniowej kinetyce. Nieliniowe procesy odgrywają zasadniczą rolę w tak niezwykłych zjawiskach, jak reakcje oscylacyjne oraz inne procesy samoorganizacji. Na tę właściwość będę się powoływał w przyszłości przy opisie przyczyn i mechanizmu reakcji oscylacyjnych.
dwa reduktory: „szybki” i „powolny”
Proponowane dalej warianty wymagają użycia dość stężonych roztworów substratów. Ilości powstałego jodu są tak duże, że trudno jest obserwować typowy niebieski kolor kompleksu: mieszanina jest niemal czarna od strąconej zawiesiny jodu. Tu proponuję użycie znacznie bardziej rozcieńczonych roztworów, co umożliwi zaobserwowanie niebieskiego koloru kompleksu jodu ze skrobią. Oprócz siarczynu zostanie także użyty drugi reduktor (kwas malonowy) tak dobrany, aby niemal nie reagował z jodanem. Przebieg reakcji jest więc początkowo identyczny, jak w klasycznej wersji. Powstały jod jest jednak powoli redukowany przez kwas malonowy - do bezbarwnego jodku. Po raptownym zabarwieniu na niebiesko, kolor ten powoli zanika, i po kilkudziesięciu sekundach roztwór ponownie staje się bezbarwny.
roztwór A: 2 g jodanu potasu w litrze wody
roztwór B: 0,4 g pirosiarczynu +
10 g kwasu malonowego + skrobia (roztwór nie nadaje się do dłuższego
przechowywania).
Zmieszać jednakowe objętości roztworów A i B. Po kilkunastu sekundach bardzo raptownie zawartość naczynia staje się ciemnoniebieska, a następnie stopniowo barwa ta zanika w ciągu kilkudziesięciu sekund.

a gdyby nadmiar reduktora?
Jaki byłby efekt po zmieszaniu jodanu z nadmiarem siarczynu? Oczywiście wszystkie procesy będą biegły identycznie jak poprzednio. Po skonsumowaniu całej ilości jodanu pozostaje jednak nadmiar siarczynu, który nie pozwoli na powstanie wolnego jodu tworzącego ze skrobią niebieskie zabarwienie. Ostatecznym wynikiem zmieszania bezbarwnych roztworów reagentów, jest bezbarwny roztwór jodku. Oczywiście doświadczenie takie ma nikłą wartość demonstracyjną... Można jednak zapytać audytorium: jak udowodnić, że w roztworze po zmieszaniu bezbarwnych roztworów zachodzą w ogóle jakiekolwiek procesy (powstawanie jodków)? Do śledzenia postępu procesów można wykorzystać sole rtęci (a również srebra). Z anionami jodkowymi dają one czerwony (w stanie koloidalnym: pomarańczowy) osad jodku rtęci HgI2 .
W zlewce zmieszać: 50 ml roztworu jodanu, 300 ml wody, 100 ml roztworu soli rtęci oraz 100 ml roztworu siarczynu. Po ok. 20 sekundach powstaje pomarańczowy osad.

niedomiar soli rtęci, nadmiar reduktora
W zlewce zmieszać: 50 ml roztworu jodanu, 300 ml wody, 45 ml roztworu soli rtęci oraz 100 ml roztworu siarczynu. Po ok. 15 sekundach powstaje pomarańczowy osad, który jednak po dalszych kilkunastu sekundach rozpuszcza się, dając bezbarwny roztwór jodortęcianu:
(czerwony) HgI2 + 2 I– → HgI42– (bez wny)
Reakcja Landolta pełni rolę laboranta dodającego jony jodkowe do roztworu soli rtęci: najpierw powoli, potem coraz szybciej...
nadmiar soli rtęci, nadmiar jodanu ("Old Nassau Reaction")
W zlewce zmieszać: 50 ml roztworu jodanu, 300 ml wody, 60 ml roztworu soli rtęci, oraz 50 ml roztworu siarczynu. Po ok. 20 sekundach powstaje stopniowo pomarańczowy osad, a po ok. 1 minucie roztwór raptownie staje się czarny. Sole rtęci wiążą powstające aniony jodkowe, które są katalizatorem reakcji Landolta i w ten sposób opóźniają jej przebieg. Taki jest typowy mechanizm działania inhibitorów: są to substancje usuwające katalizator ze środowiska reakcji. Im więcej w roztworze znajduje się soli rtęci, tym później powstaje ciemne zabarwienie. Tę modyfikację odkryli studenci jednego z uniwersytetów w USA; w literaturze znana jest ona pod nazwą "Old Nassau Reaction".
niedomiar soli rtęci, nadmiar jodanu
W zlewce zmieszać: 50 ml roztworu jodanu, 300 ml wody, 45 ml roztworu soli rtęci oraz 50 ml roztworu siarczynu. Po kilkudziesięciu sekundach powstaje pomarańczowy osad, który po dalszych kilku sekundach rozpuszcza się. Moment później zawartość zlewki raptownie staje się czarna.
wskaźnik pH, nadmiar reduktora
Później zostanie wykazane, że sumaryczne równanie reakcji Landolta, ma postać:
IO3– + 3 HSO3– → I– + 3 SO42– + 3 H+
Postępowi reakcji Landolta towarzyszy więc stopniowe zakwaszanie roztworu (później wykażemy, że jest to już drugi mechanizm autokatalizy). Zmiany te można wykazać za pomocą wskaźnika pH: błękitu bromofenolowego (w środowisku powyżej pH 4,6 jest on purpurowoniebieski, a poniżej pH 3,0 - żółty). Wbrew pozorom, da się zastosować do tego tylko nieliczne inne wskaźniki, np. czerwień Kongo (pH 5,3 - 3,0). Dodatek NaOH ma na celu zwolnienie tempa przemian i przesunięcie pH w zakres, który pozwoli na wyraźniejsze zaobserwowanie zmian.
W zlewce zmieszać: 50 ml roztworu jodanu, 200 ml wody, 6 ml roztworu NaOH, kilka kropel wskaźnika, oraz 100 ml roztworu siarczynu. Po kilkunastu sekundach fioletowa barwa roztworu przechodzi w zielononiebieską, a potem w żółtą. Jeśli czas zmiany barwy wskaźnika jest zbyt długi, można zmniejszyć ilość NaOH. Można powtórzyć tę wersję z czerwienią Kongo (lub błękitem tymolowym ?).

Jeden z Czytelników (Grzegorz M.) zaproponował:
Przeprowadziłem reakcje zgodnie z podanymi proporcjami - zastępując błękit - oranżem. Reakcja ma identyczny przebieg - zarówno przy nadmiarze reduktora jak i nadmiarze jodanu. Zmiana żółtopomarańczowego roztworu w czerwony nie jest aż tak widowiskowa jak niebieski- żółty dla błękitu - ale popularność oranżu aż nadto wynagradza tę wadę.
Poza tym płynne ściemnianie roztworu ma tu także sporo wartości naukowych - których przy błękicie nie można zaobserwować.
Ciekawy efekt daje też użycie czerwieni metylowej, co prawda zakres zmiany barwy jest znacznie wyżej (4,4-6,2), przez co zmiany barwy nie pokrywają się z końcem reakcji niemniej jednak efekty wizualne i naukowe są też ciekawe. Wymaga to użycia delikatnie większej ilości zasady na wstępie- tak aby dopasować pH roztworu do wskaźnika.
Przy reakcji z nadmiarem reduktora efekt podobny - zmiana z żółtej barwy przez różową do czerwonej.Przy nadmiarze jodanu początkowy etap jest taki sam - ale wydzielenie jodu a zarazem powstanie bardzo ciemnej barwy kompleksu ze skrobią jest znacznie oddalone czasowo od zmiany barwy wskaźnika. Co jest zrozumiałe ze względu na znaczną różnice wartości pH zmiany barwy czerwieni metylowej, a powstania jodu.
wskaźnik pH, nadmiar jodanu
W zlewce zmieszać: 50 ml roztworu jodanu, 200 ml wody, 6 ml roztworu NaOH oraz 50 ml roztworu siarczynu. Po ok. 30 sekundach fioletowa barwa roztworu przechodzi w zielononiebieską a potem w żółtą, i w kilka sekund później, bardzo raptownie - w czarną.
** Landolt w UV
Poszczególne reakcje lekko zaalkalizowanego roztworu prowadzić można z użyciem niewielkiego dodatku chininy, pełniącej tu rolę wskaźnika pH (w UV po zakwaszeniu, sprotonowana forma chininy fluoryzuje w świetle lampy UV. **
wskaźnik pH, nadmiar soli rtęci, nadmiar reduktora
W zlewce zmieszać: 200 ml wody, 100 ml roztworu siarczynu, 100 ml roztworu soli rtęci, kilka kropel wskaźnika, 17 ml roztworu NaOH oraz na koniec - 50 ml roztworu jodanu. Po ok. 15 sekundach fioletowa barwa przechodzi stopniowo w zielononiebieską i żółtą, a po dalszych 20 sekundach rozpoczyna się strącanie pomarańczowego osadu. Zmieszanie soli rtęci z roztworem wodorosiarczynu powoduje niespodziewanie silne zakwaszenie. Możliwe, że powstaje niezbyt trwały kompleks siarczynowy z jonami rtęci, a uwolnione jony wodorowe powodują to dodatkowe zakwaszenie. Dlatego w tej wersji reakcji lepiej jodan dodawać dopiero na końcu.
wskaźnik pH, niedomiar soli rtęci, nadmiar reduktora
W zlewce zmieszać: 200 ml wody, 100 ml roztworu siarczynu, 20 ml roztworu soli rtęci, kilka kropel wskaźnika, 6 ml roztworu NaOH oraz na koniec - 50 ml roztworu jodanu. Po kilku sekundach fioletowa barwa przechodzi stopniowo w zielononiebieską i żółtą, i w moment później rozpoczyna się strącanie pomarańczowego osadu, który po dalszych kilkunastu sekundach rozpuszcza się dając klarowny, ale żółty roztwór.
wskaźnik pH, nadmiar soli rtęci, nadmiar jodanu
W zlewce zmieszać: 200 ml wody, 50 ml roztworu siarczynu, 60 ml roztworu soli rtęci, kilka kropel wskaźnika, 9 ml roztworu NaOH oraz na koniec - 50 ml roztworu jodanu. Po kilkunastu sekundach fioletowa barwa przechodzi stopniowo w zielononiebieską i żółtą, i w moment później rozpoczyna się strącanie pomarańczowego osadu, a po dalszych kilkudziesięciu sekundach powstaje raptownie ciemne zabarwienie.
wskaźnik pH, niedomiar soli rtęci, nadmiar jodanu
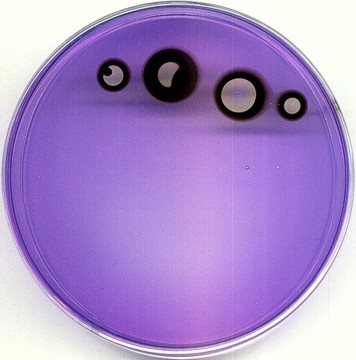
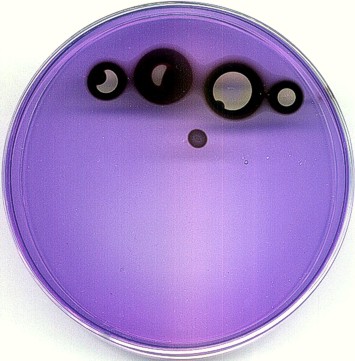
W zlewce zmieszać: 225 ml wody, 50 ml roztworu siarczynu, 15 ml roztworu soli rtęci, kilka kropel wskaźnika, 3 ml roztworu NaOH oraz na koniec - 50 ml roztworu jodanu. Po kilkunastu sekundach fioletowa barwa przechodzi stopniowo w zielononiebieską i żółtą, w chwilę później rozpoczyna się strącanie pomarańczowego osadu, który po dalszych kilkunastu sekundach rozpuszcza się dając żółty klarowny roztwór. Dosłownie w sekundę później powstaje raptownie ciemne zabarwienie. Ta pięciobarwna wersja jest w praktyce trudna do wykonania i wymaga wielu wstępnych prób. Należy zgrać ze sobą zarówno rozcieńczenie roztworu, jak i początkową kwasowość roztworu, a również ilość soli rtęci (zbyt mała ilość może w ogóle nie wytrącać osadu, a zbyt duża sprawia, że osad nie zdąży się rozpuścić).
Ponieważ czas pomiędzy rozpuszczeniem się pomarańczowego osadu HgI2, a powstaniem jodu jest bardzo krótki, na czwartym miejscu tego szeregu fotografii musiałem umieścić drugi skan powtórnie...
przepowiadanie przyszłości (być w roli wróżbity)...
Duże wrażenie na widzach robi umiejętność przepowiedzenia efektu końcowego reakcji zegarowej zwłaszcza, gdy nastawić reakcję Landolta tak, aby efekt końcowy wystąpił dopiero po kilku minutach. Posłużyć się można do tego obserwacją z bliska papierka wskaźnikowego, który wcześniej został przylepiony taśmą klejącą wewnątrz zlewki. Skrawek powinien być złożony w kilka warstw i taśma klejąca powinna utrudniać wypłukiwanie barwnika przez mieszany roztwór. W praktyce trzeba tu użyć "zwężonych" papierków dobrej jakości, o odpowiednio dobranym zakresie zmian pH. Można też stanąć tyłem do zlewki z reagującym bezbarwnym roztworem, pobrać do małej zleweczki jego porcję, dodać do niej niewielką ilość wskaźnika i obserwować zmiany barwy poprzedzające efekt końcowy.
Trudno jest jednak o precyzyjną kontrolę pH. O wiele lepsze wyniki daje włożenie do naczynia z reagującym roztworem, sondy pH-metru. Początkowa wartość pH wynosi 4 - 5 (zależy ona oczywiście od proporcji substratów oraz stopnia rozcieńczenia). Zgodnie z równaniem:
IO3– + 3 HSO3– (słaby kwas) → I– + 3 SO42– + 3 H+ (mocny kwas)
przebiegowi reakcji towarzyszy spadek pH. pH-metr daje doskonałą możliwość kontroli tych zmian. Doświadczenie wskazuje, że wydzielenie się jodu (ciemne zabarwienie) następuje po osiągnięciu pH ok. 2,2. Jeśli reakcję uruchomić w dostatecznie dużym rozcieńczeniu, to daje to możliwość bardzo precyzyjnego śledzenia zmian pH i uprzedzenia momentu końcowego o 2-3 sekundy - i to przy całkowitym czasie trwania reakcji około 2-3 minut! (nie należy jednak przesadzać z tym zwlekaniem - reakcja może okazać się szybsza i stracimy efekt wraz z własną reputacją wobec audytorium...). Osoby mające zdolności elektrotechniczne mogą zbudować prosty automat ostrzegający dzwonkiem o zbliżaniu się efektu końcowego (jest to nieco ryzykowne: ktoś może zarzucić, że czynnikiem wyzwalającym powstanie jodu - jest właśnie dźwięk dzwonka. Podobnie jak mieszanie herbaty łyżeczką powoduje, że jest ona słodka...).
dokładniej o zmianach pH
Poniżej przytaczam wykres zmian pH zarejestrowanych podczas klasycznej wersji reakcji Landolta. Przy interpretacji krzywej koniecznie trzeba pamiętać, że przedstawia ona co prawda zmiany stężenia jednego z produktów (H+), ale że wartości liczbowe podane są w skali logarytmicznej (pH); zmiany kwasowości roztworu są więc w rzeczywistości o wiele gwałtowniejsze, niż wydaje się to na pierwszy rzut oka (spadek pH o 1 jednostkę pH odpowiada w rzeczywistości dziesięciokrotnemu wzrostowi stężenia kwasu; dwie jednostki odpowiadają stukrotnej zmianie stężenia kwasu!).
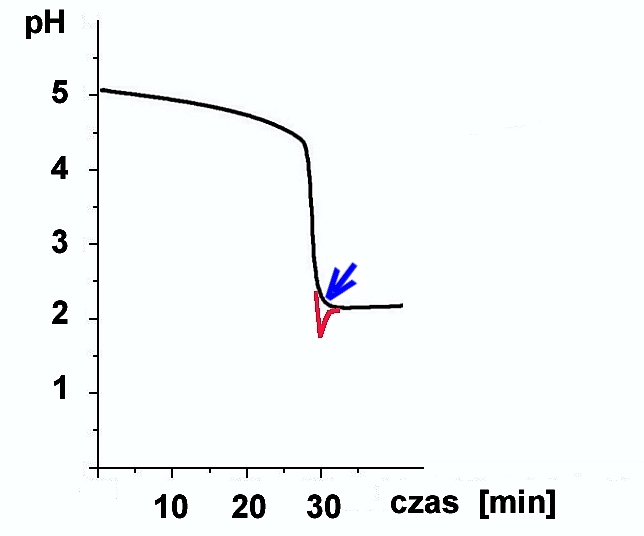
Wykonanie tych pomiarów okazało się nadspodziewanie kłopotliwe; czas odpowiedzi pomiarowej elektrody szklanej był tak długi (1 - 2 minuty!), że uniemożliwiało to "nadążanie" odczytu za szybkim tempem reakcji. Ostatecznie trzeba było wydłużyć czas reakcji do ok 30 minut - przez odpowiedni dodatek NaOH w momencie początkowym reakcji (patrz niżej).
Charakterystyczny jest niewielki wzrost pH po rozpoczęciu wydzielania się jodu (ten moment zaznaczono na wykresie niebieską strzałką). Jest to doskonale widoczne na skali pH-metru, jednak na wykresie nie zostało zarejestrowane z powodów wyjaśnionych wyżej (dorysowano jednak ten fragment ręcznie na czerwono). Reagująca mieszanina jodków i jodanu zachowuje się tak jak zasada Brönsteda (reaguje z jonami wodorowymi), pomimo, że żaden z jej składników nie ma wyraźniejszych właściwości zasadowych. Jest to bardzo interesujące rozszerzenie klasyfikacji zasad Brönsteda, mające zresztą praktyczne zastosowanie w chemii analitycznej.
Ponieważ znana jest powszechna niechęć do logarytmów, zatem jeszcze krótki komentarz do wykresu. Gdyby obrazem zmian była prosta o jednostajnym nachyleniu, w rzeczywistości oznaczałoby to gwałtownie przyspieszające tempo zakwaszania. Takie są bowiem konsekwencje skali logarytmicznej (na osi rzędnych nie są odkładane wartości stężenia, ale wartości logarytmów stężenia: pH). W doświadczeniu tym jednak krzywa ma gwałtownie malejący przebieg. W przeliczeniu na stężenie kwasu odpowiada to niewiarygodnie przyspieszające tempo zmian szybkości reakcji. W ostatniej, trzydziestej minucie reakcji, tempo zmian jest zapewne wiele tysięcy razy większe niż w ciągu całego poprzedniego okresu. Taki jest rezultat podwójnej autokatalizy w Reakcji Landolta... Wygląd wykresu do złudzenia przypomina krzywą miareczkowania, chociaż w tym wypadku nie tylko nie dodawano titranta - ale to właśnie biegnąca reakcja sama wytwarzała jony wodorowe. Ponieważ pomiary wykonywano za pomocą autotitratora (z odłączonym dozownikiem), zatem powyższą zależność można by nazwać krzywą autoautotitrowania...
Proponuję jeszcze jedną, efekciarską wersję pH-metryczną. Należy zaproponować komuś z audytorium, aby uruchomił jedną z przygotowanych wersji reakcji Landolta: jedną z nadmiarem jodanu, lub drugą - z nadmiarem siarczynu. Stojąc tyłem do reagującego roztworu, należy rozstrzygnąć: którą wersję uruchomiono? Pomocą będzie obserwacja skali pH-metru: czy występuje niewielki wzrost pH po pierwotnym stopniowym spadku pH do minimalnej wartości ok. 2,2 (moment zużycia jednego z substratów).
powoli, jeszcze wolniej, bardzo bardzo powoli...
Jeśli zamierza się pokazać reakcję Landolta tak, aby czas niezbędny do powstania ciemnego zabarwienia jodu był możliwie długi, wystarczy odpowiednio rozcieńczyć mieszaninę. Co prawda czas reakcji zostaje w ten sposób przedłużony, ale faza ciemnienia roztworu staje się znacznie "rozmyta" w czasie. O wiele lepsze wyniki daje wstępne zaalkalizowanie (a właściwie: zmniejszenie kwasowości) mieszaniny. Co więcej, obserwuje się wtedy interesujące efekty dodatkowe. Przygotować szereg zleweczek, w których umieścić po 15 ml wody, po 5 ml roztworu wodorosiarczynu, następnie dodać podane niżej objętości 0,2 M NaOH, oraz na koniec, po 5 ml roztworu jodanu; roztwory mieszać. Można w ten sposób uzyskać okresy indukcji nawet powyżej godziny, przy zachowaniu nadspodziewanie dużej raptowności ostatecznego efektu.
ml 0,2 M NaOH |
czas |
0 |
11 s |
0,5 |
48 s |
1,0 |
1 min. 29 s |
1,5 |
3 min. 29 s |
2,0 |
ok. 9 min. |
2,5 |
ok. 25 min. |
3,0 |
ok. 1,5 godz., dopiero po ogrzaniu |
3,5 |
nie powstaje |
Pomimo tak ogromnie wydłużonego czasu, efekt wizualny związany z wydzieleniem jodu jest niemal tak samo raptowny, jak w przypadku pierwszego roztworu! Jest to o tyle zrozumiałe, gdyż reakcja przebiega w warunkach w których stężenie tylko jednego z reagentów (jonów wodorowych) jest bardzo znacznie obniżone, przy dużych stężeniach pozostałych substratów [porównaj równanie kinetyczne (4)]. W tym miejscu nasuwa się pytanie: czy jest w ogóle taka początkowa wartość pH, powyżej której reakcja Landolta w ogóle nie zachodzi? W przypadku dwóch ostatnich roztworów spodziewane czasy byłyby rzędu godzin... Dla ewentualnego przyspieszenia ich biegu, po godzinie bezskutecznego oczekiwania wstawiono więc zlewki do łaźni wodnej o temperaturze 60 stopni, co daje ponad 10-krotne przyspieszenie procesów (ostatni roztwór dla pewności pozostawiono jeszcze na tydzień w szczelnie zamkniętej kolbce. Ciemne zabarwienie jednak nie powstało). Tak więc wydaje się, że rzeczywiście istnieje wartość progowa początkowego pH mieszaniny, powyżej której reakcja Landolta w ogóle nie zachodzi. Ten efekt zostanie wykorzystany nieco dalej, przy pokazie reakcji z udziałem kationitu.
** Można zaprezentować efekciarski pokaz szeregu naczyń w których powstanie barwy (lub wielu kolejnych barw) następuje kolejno, z kilkusekundowym przesunięciem w xczasie. Można oczywiście kolejne roztwory nieco rozcieńczać wodą, ale można też efekt opóźnienia uzyskać niewielkim dodatkiem roztworu NaOH: http://www.youtube.com/watch?v=Tv6_IsdnaGg **
szybciej, szybciej, jeszcze szybciej...
Propozycja pokazu w postaci serii roztworów kolejno zmieniających barwę. Przygotować np. 500 ml reagenta z dodatkiem odpowiedniej ilości zasady (V ml 0,4 M NaOH; sposób wyznaczenia ilości NaOH blokującej reakcję podano wcześniej). Roztworem takim można bez pośpiechu manipulować, bo bieg reakcji jest praktycznie wstrzymany. Jej postęp uruchamia się i przyspiesza przez stopniowe dodawanie coraz większych ilości kwasu. Do 5 pustych zlewek dodać kolejno: 0,2V ml; 0,4V ml; 0,6V ml; 0,8V ml; 1,0V ml 0,4M HCl. Do każdej zlewki wlać po 100 ml reagenta. Czas reakcji zależy od ilości kwasu solnego.
zawsze od góry; samoorganizacja w reakcji Landolta?...
Przygotować dokładnie (ale szybko) wymieszany roztwór z poprzedniej serii zaalkalizowany tak, aby czas reakcji wynosił ok. 10 minut i pozostawić go bez mieszania. Pod koniec biegu reakcji pojawiają się tuż przy górnej powierzchni, przy wewnętrznych ściankach zlewki, regularne rozszerzające się ciemniejące plamy, które po pewnym czasie rozszerzają się na całą objętość naczynia. Ich pojawianie się w górnej części wynika prawdopodobnie w faktu nierównomierności temperatury oraz konwekcji towarzyszącej efektowi cieplnemu reakcji Landolta (u góry znajduje się nieco cieplejsza strefa, w której reakcja jest szybsza). Natomiast regularność kształtu nasuwa przypuszczenie o powstaniu stref samoorganizacji, podobnie nieco jak przy zjawisku Bénarda. Jeśli w takim momencie roztwór jednorazowo zamieszać, barwa zanika. Natomiast po pewnym czasie na powierzchni tworzy się kolisty ciemno zabarwiony pierścień o średnicy zbliżonej do połowy średnicy naczynia, który stopniowo rozszerza się zarówno do środka, jak i ku ściankom naczynia. Być może pochodzi on ze zmieszania lokalnych brzegowych miejsc samoorganizacji. Czyżby pomimo zamieszania, fragmenty roztworu zachowały "pamięć" tych zarodków samoorganizacji?

wersja magiczna z prawą dłonią...
Przygotować mieszaninę: 50 ml roztworu jodanu, 500 ml wody, 50 ml wodorosiarczynu. Po wymieszaniu reagujący roztwór rozlać do dwóch zlewek. Jedną z nich zachować do kontroli i odsunąć ją w bok, a drugą "napromieniowywać bioenergią", za pomocą prawej dłoni rozpostartej nad jedną ze zlewek.
ten magiczny gest można podpatrzyć w różnych popularnych programach serwowanych w TV, w których np. "magnetyzuje się" wodę w celu nadania jej cudownych właściwości leczniczych. To nie jest żart! Przeglądnij jeszcze dziś program TV; takie pozycje rzeczywiście nadawane są w polskiej telewizji publicznej...
W zlewce "napromieniowanej" prawą dłonią ciemne zabarwienie powstaje znacznie szybciej, niż w roztworze kontrolnym!

Tajemnica tego efektu jest prosta: do jednej ze zlewek (trzeba koniecznie pamiętać: której!) dodaje się ukradkiem przed pokazem, kroplę dość stężonego roztworu KI (nie za dużo, kilkanaście mg).
Audytorium nie należy jednak zbyt długo trzymać w nieświadomości; ludzie są bowiem zadziwiająco skłonni do wierzenia w każde bzdury. Dowodem niech będzie popularność takich właśnie programów TV, a również poczytność pism w rodzaju "Wróżka", horoskopów itd., a wreszcie wyniki różnych sprawdzianów społecznych...
Doświadczenie to ma jednak swoją wartość dydaktyczną, jest bowiem dowodem katalitycznych właściwości anionów jodkowych! Roztwór "napromieniowywany" już od początku zawierał dodatek substancji będącej katalizatorem, natomiast reakcja w roztworze kontrolnym przebiegała początkowo bardzo powoli; dopiero po pewnym czasie powstawała substancja przyspieszająca bieg reakcji. W identyczny sposób (chociaż już bez magii dłonią), za pomocą dodatku jakiegokolwiek kwasu, można wykazać istnienie drugiego mechanizmu autokatalizy w reakcji Landolta.
wersja magiczna z lewą dłonią...
Gdy wyjaśni się widzom sposób oszustwa w poprzednim pokazie, z reguły audytorium reaguje nonszalancko: "takie to było proste!". W tej sytuacji daję widzom do przemyślenia problem: jak zaaranżować podobny pokaz, który jednak miałby na celu próbę wmówienia, że "napromieniowywanie" lewą dłonią, powoduje opóźnienie efektu końcowego reakcji Landolta? (różni szarlatani nie wahaliby się tu użyć terminu "ujemna bioenergia"). Dla utrudnienia: należy w tym celu posłużyć się również ukradkowym dodatkiem jodku potasu, który jednak reakcję Landolta przyspiesza, a nie opóźnia...

z kationitem
Przygotować mieszaninę: 5 ml roztworu jodanu, 60 ml wody, 5 ml wodorosiarczynu. Mieszaninę szybko wlać do płytki Petriego tak, aby warstwa cieczy nie była grubsza niż ok. 3 mm, jednocześnie do roztworu ostrożnie wrzucić kilkanaście większych ziarenek dowolnego F kationitu sulfonowego w formie kwasowej. Dla zmniejszenia rozmywania stref zalecam użycie roztworów z dodatkiem zawiesiny prawie przezroczystego żelu krzemionkowego do chromatografii (lub lżejszej sproszkowanej celulozy).
Jeśli dysponuje się agarem, to o wiele lepsze wyniki daje reakcja przebiegająca w środowisku żelowym; w ostateczności można użyć gęstego kisielu skrobiowego (patrz nieco dalej).
Po kilkudziesięciu sekundach roztwór wokół ziaren kationitu ciemnieje, a ciemne strefy stopniowo się rozszerzają. Z reguły widoczne są smugi spowodowane przez przypadkowe ruchy cieczy (a również różnice temperatur), wreszcie ciemne zabarwienie raptownie rozszerza się na całe naczynie. Trochę przypomina to obserwację w przyspieszonym tempie wzrostu hodowli kolonii bakteryjnych.
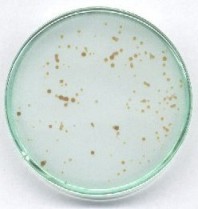
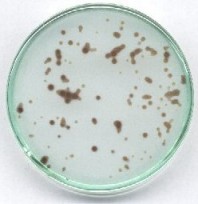
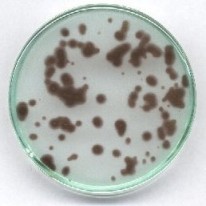

W najbliższym sąsiedztwie ziaren zachodzi wymiana jonowa kationów soli i zostają uwolnione jony wodorowe. W tych silniej zakwaszonych miejscach reakcja Landolta zostaje przyspieszona i one właśnie barwią się wcześniej, niż pozostały roztwór. W ten sposób również wykazuje się katalityczną rolę jonów wodorowych (kwasu). Pozorny ruch barwnych stref wynika z różnej szybkości reakcji w miejscach o różnym zakwaszeniu. Trochę przypomina on efekt samoorganizacji w postaci wędrujących barwnych fal reaktywności chemicznej, chociaż w rzeczywistości są to procesy zupełnie różne. Interesujące kolorystycznie są niektóre poprzednio opisane wersje - dodatkowo z użyciem jonitu (szczególnie przy niedomiarze soli rtęci i nadmiarze reduktora).
Podczas wykonywania tej wersji doświadczenia trzeba się spieszyć, aby reakcja Landolta nie zakończyła się zbyt szybko. O wiele łatwiej wykonać to z mieszaniną, którą wstępnie zaalkalizowano roztworem NaOH tak znacznie, że uniemożliwia to w ogóle jej przebieg (patrz nieco wcześniej: "powoli..."). Taki roztwór z dodatkiem żelu krzemionkowego wlać cienką warstwą na płytkę Petriego i dodać kilka ziaren kationitu. Powstaną rozszerzające się strefy ciemnego zabarwienia, jednak nie rozciągną się one na całą powierzchnię naczynia.
z anionitem
Również widowiskowy jest przebieg doświadczenia z ziarnami F anionitu w formie zasadowej (silnie lub słabo zasadowego). Reakcja Landolta w otoczeniu ziaren zostaje opóźniona na skutek zaalkalizowania. Cały roztwór po pewnym czasie staje się ciemny, za wyjątkiem bezbarwnych plam wokół ziaren anionitu. Zamiast zasadowego anionitu można użyć kryształu jakiejkolwiek niezbyt łatworozpuszczalnej soli o odczynie zasadowym, np.: NaHCO3. Czytelnikowi pozostawiam przewidzenie efektu zastosowania anionitu w formie jodkowej...
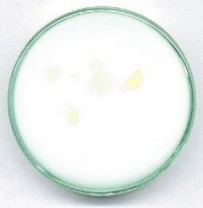
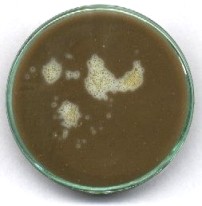
"Landolt w galarecie"
Wiele interesujących zjawisk można obserwować swobodnie tylko wtedy, gdy reagujący roztwór jest zupełnie nieruchomy. Wcześniej proponowano w tym celu dodatek stałego wypełniacza w rodzaju żelu krzemionkowego. O wiele bardziej eleganckie i skuteczne jest użycie środka żelującego: agaru. Ponieważ roztwory należy mieszać w temperaturze ok. 30 stopni, skład roztworów powinien taki, aby czas reakcji był długi - kilka godzin (dodatek NaOH). Zamiast wody należy po prostu użyć ok. 3-procentowego roztworu agaru we wrzącej wodzie. Po zmieszaniu z pozostałymi roztworami, mieszaninę ochłodzić szybko do ok. 30 stopni, dodać teraz dopiero roztwór ostatniego reagenta i mieszaninę szybko wlać do płytki Petriego ustawionej na zimnym (najlepiej: metalowym) podłożu. Czas krzepnięcia nie przekracza jednej minuty. Przykładowo, można użyć roztworu: 15 ml wody (roztworu agaru), 5 ml roztworu wodorosiarczynu, 2-2,5 ml 0,2 M NaOH, oraz na koniec, 5 ml roztworu jodanu. Po zastygnięciu, na powierzchni żelu umieścić kilka ziarenek silnie kwaśnego kationitu sulfonowego. Powstają regularne, rozszerzające się strefy szybszego zachodzenia reakcji Landolta; rosną one również w kierunku dna naczynia. Po dłuższym czasie cała masa żelu raptownie przyjmuje ciemne zabarwienie. Można temu zapobiec stosując silniej zaalkalizowany roztwór; dodając nie 2-2,5, lecz 4-5 ml NaOH.
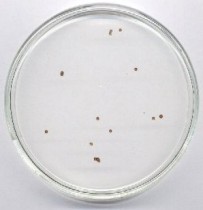
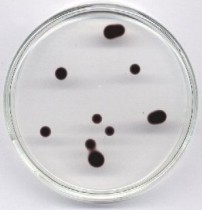
Wersje „w galarecie” można mnożyć, np. pomarańczowy rozszerzajacy się pierścień - blokująca reakcję ilość NaOH, nadmiar NaHSO3, niedomiar soli rtęci, ziarno kationitu; ew. dodatek chininy lub błękitu bromokrezolowego.
W eksperymentach w agarze istotną rolę odgrywa grubość warstwy żelowej. Strefa produktów reakcji rozszerza się jednolicie wokół ziarna reagenta umieszczonego na powierzchni, w rezultacie ma ona kształt półkuli. Widać to doskonale, gdy kryształek umieścimy blisko bocznej ścianki i z boku obserwujemy wynik. Powoduje to jednak zmieniającą się stopniowo intensywność barwy, gdy plamę obserwujemy z góry (np. na rzutniku pisma). Trzeba często poczekać dostatecznie długo, aby strefa produktów przedyfundowała przez całą miąższość warstwy żelu. Czasami efekt jest bardzo interesujący. Ale lepiej jest prowadzić reakcję w warstwach o grubości nie większej ni 2 mm. Należy więc użyć wystarczająco dużego stężenia wskaźników, aby pomimo to barwa była wyraźna.
"zaćmienie księżyca w galarecie"
Przygotować wersję reakcji z nadmiarem jodanu, zablokowanej odpowiednio dużym dodatkiem NaOH. Zamiast wody użyć 2-procentowego ciepłego roztworu agaru. Ciepłą mieszaninę wlać na płytkę Petriego i pozostawić do zastygnięcia. Na powierzchni żelu umieścić dwa ziarna jonitów: kwasowego kationitu i silnie zasadowego anionitu; ziarna umieścić w odległości kilku mm od siebie. Zamiast kwasowego kationitu użyć można kryształka kwasu szczawiowego, a zamiast zasadowego anionitu, większy kryształ uwodnionego węglanu sodu. Po krótkim czasie wokół kationitu powstaje rozszerzająca się ciemna strefa jodu. Z chwilą gdy granica frontu wejdzie w kolizję z niewidoczną, również rozszerzającą się strefą zasadowości wokół anionitu, ciemna plama będzie przybierała kształt podobny do księżyca podczas zaćmienia.
Efekty po naniesieniu małej porcji wodorosiarczynu na warstwę agaru są tak zaskakujące, że wymaga to dokładniejszego opisu. Jak to możliwe, aby dodatek reduktora powodował jednak zajście reakcji utleniania; skąd bierze się podwójny obraz ciemnej otoczki z żółtym środkiem; dlaczego wreszcie żółta strefa najpierw się rozszerza, a potem kurczy? I dlaczego ostatecznym efektem naniesienia reduktora jest jednak zajście utlenienia?
Warstwa agarowa zawierała nadmiar jodanu oraz wskaźnik, a bieg reakcji został zablokowany dość dużym dodatkiem NaOH (fioletowy kolor agaru).
Wodorosiarczyn jest zarówno reduktorem, jak i substancją wyraźnie kwasową. Po naniesieniu odrobiny stałego NaHSO3 na warstwę żelu, szybciej dyfundujące jony wodorowe zakwaszają najbliższe sąsiedztwo, co powoduje powstanie ciemnej plamki jodu. W wyniku reakcji Landolta jony wodorowe zostają powielane i dyfundują na zewnątrz powodując rozszerzanie się ciemnej strefy. Ale także wolniej dyfundujące redukujące jony wodorosiarczynowe penetrują warstwę żelu, powodując powstawanie rozszerzającej się żółtej strefy redukcji w środku plamy. W odróżnieniu od namnażających się jonów wodorowych, siarczyn w miarę rozprzestrzeniania się zostaje zużywany. I dlatego środkowa żółta strefa redukcji co prawda rozszerza się, ale zaczyna to być coraz powolniejsze. A w dalszym otoczeniu żel zawiera bardzo znaczny nadmiar jodanu, który powoli także dyfunduje - z zewnątrz do środka. I stopniowo żółta strefa redukcji zaczyna się cofać i kurczyć. Natomiast ciemna strefa jodu długo jeszcze się rozszerza.
To trochę jak analogia z wysadzeniem silnego, ale niewielkiego desantu wojskowego na nieprzyjacielskim terenie umiarkowanie nasyconym siłami wojskowymi. Po lądowaniu zaczyna się ekspansja desantu, jako skutek lokalnej jego przewagi zbrojnej. W miarę ekspansji, przewaga ta staje się coraz mniej dominująca. Poza tym desant ponosi ciągłe ubytki na skutek walki z kontratakiem, a siły nieprzyjacielskie dysponują jednak znaczną globalną przewagą. Co prawda rozproszoną na większym obszarze, ale jednak to ich przewaga powoduje po dłuższym czasie zahamowanie ekspansji i zlikwidowanie desantu. Po zakończeniu walki pozostają jednak w terenie resztki mało uchwytnych ruchliwych dywersantów, którzy skutecznie zajmują się werbunkiem pomocników. Ten złożony obraz procesów chemicznych i dyfuzji fizycznej może być pomocny przy zrozumieniu zjawisk wędrujących fal reaktywności chemicznej.
Zachęcam do wypróbowania innych odczynników w agarze. Kwas szczawiowy i cytrynowy dają okrągłą plamę jodu. Kwas malonowy daje ciemną plamę jodu, ale po pewnym czasie w środku powstaje jasna plama produktów wtórnej redukcji jodu przez nadmiar redukującego kwasu malonowego.
Wodorosiarczyn daje wynik zaskakujący, bo powoduje najpierw powstanie ciemnej plamy jodu (decydujący jest efekt katalizy po zakwaszeniu; jest jednak paradoksem, że dodatek REDUKTORA powoduje efekt UTLENIENIA); dopiero potem w środku pojawia się jaśniejsza plama produktów redukcji nadmiarem wodorosiarczynu.
Należy poczekać dość długo, aby wewnętrzna jasna strefa redukcji dotarła do dna naczynia; dopiero wtedy jest ona zupełnie żółta i przezroczysta. Jeśli w niewielkiej odległości od ciemnej plamy, po zewnętrznej jej stronie umieścić kryształek tiosiarczanu lub siarczynu, to na krawędzi rosnącej ciemnej plamy powstaje półkolisty przezroczysty wżer redukcji.
Najciekawszy efekt dają eksperymenty z naniesieniem wodorosiarczynu i jodanu. Po pewnym czasie tak jak poprzednio powstaje rozszerzający się, nieco rozmyty ciemny pierścień jodu, a wewnątrz niego zupełnie przezroczysty żółtawy krążek strefy redukcji, o bardzo ostrych krawędziach. Jeśli na ciemnym pierścieniu umieścić kryształ jodanu, to po pewnym czasie uzyskuje się efekt łudząco podobny do zaćmienia słońca: na krawędzi jasnożółtej tarczy powstaje półkolista ciemna plama.
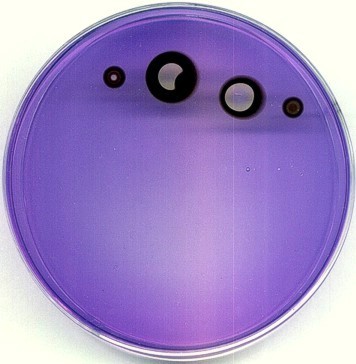
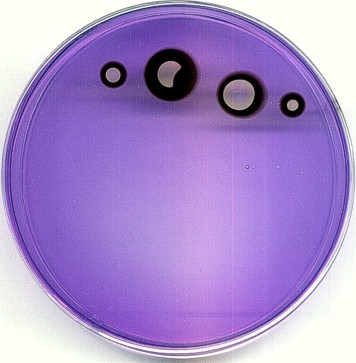
Efekt ciemnego "wżeru" uzyskano nakładając nieco z boku, na ciemny pierścień - kryształka jodanu; naniesienie go na niebieskie podłoże nie powoduje żadnego efektu, bo przebieg reakcji jest tam zablokowany. Można także uzyskać efekt, jakiego nie zobaczą nigdy astronomowie: tarczę z dwiema plamami...
Jako ciekawostkę załączam link do zdjęcia potrójnego zaćmienia Jowisza przez jego księżyce: http://gallery.astronet.pl/images/05863.jpg Prawda, że troszkę podobne? Inne: http://astronomer.republika.pl/ http://www.marcinsienko.republika.pl/pse-pol-1996-10-12.html
Można przygotować niezbyt silnie zaalkalizowane podłoże odczynnika na który nanieść kryształ kwasu malonowego lub wodorosiarczynu. Początkowo powstaje ciemna plama jodu na niebieskim tle barwy wskaźnika. Po pewnym czasie podłoże zmienia powoli kolor na zielonkawy, a następnie całość barwi się ciemno (powstaje w całej warstwie jod). Na ciemnym tle powstają przezroczyste ostro ograniczone żółte plamy. Przezroczysta plama początkowo się rozszerza, a potem stopniowo kurczy (nadmiar dyfundującego z otoczenia jodu).
Warto dokładniej obserwować końcowy efekt powstawania barwy w środowisku żelowym, są to bowiem warunki sprzyjające powstawaniu efektów samoorganizacji chemicznej. Najczęściej na styku żelu z boczną ścianką naczynia, u góry powstaje najpierw jeden, potem jeszcze kilka punktów, z których ciemna strefa rozszerza się na pozostały roztwór. Do złudzenia przypomina to typowe zachowanie "wzbudzonego" roztworu podczas reakcji F Zaikina-Winfree, podczas której powstają wędrujące kolorowe fale reaktywności chemicznej. W naszym doświadczeniu powstaje jednak tylko pojedyncza fala utlenienia (porównaj dalej: wersja "z pierścionkiem').
** Wreszcie, można uruchomić wersję z agarem oraz nadmiarem siarczynu, z solą rtęci, wszystko zalkalizowane NaOH. Powstają wtedy pierścienie koloru pomarańczowego. **
reakcja Landolta "pamięta" temperaturę
Przygotować identyczny jak przy opisie poprzedniego pokazu roztwór, z dodatkiem agaru. Mieszaninę szybko wlać na płytkę Petriego ustawioną na chłodnym podłożu. Po zastygnięciu żelu odwrócić płytkę i na szkle położyć na kilka minut ogrzaną masywną nakrętkę metalową (w miejscu A) oraz drugą, silnie oziębioną nakrętkę (w miejscu B). Ciemne zabarwienie powstaje najpierw w miejscach cieplejszych. Fragmenty ochłodzone barwią się ze znacznym opóźnieniem. Doskonale widoczny jest rozkład przypadkowych nierównomierności temperatury.
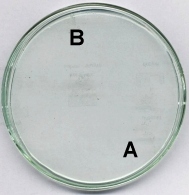

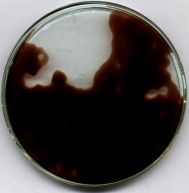

Istotna jest nie tyle temperatura w określonym miejscu płytki, co iloczyn temperatury i czasu (tak zdefiniowana całka nie ma jednak chyba swojej nazwy w fizyce).
miareczkowe wersje reakcji Landolta
W małej zleweczce (z mieszadełkiem magnetycznym) umieścić dokładnie 5,0 ml roztworu jodanu. Roztwór ten miareczkować z biurety za pomocą roztworu wodorosiarczynu; należy jednak miareczkować bardzo powoli: w tempie 1 kropla na 10 - 15 sekund. Zanotować objętość wodorosiarczynu v1 po której powstaje ciemne zabarwienie. Powtórzyć podobne miareczkowanie, dozując tym razem roztwór siarczynu bardzo szybko. Okazuje się, że wynik miareczkowania zależy głównie od tempa miareczkowania! Im szybciej się miareczkuje, tym większa jest objętość v1 (jeśli miareczkuje się dostatecznie szybko, to może się zdarzyć, że w ogóle jod się wydziela!). Raptowność pojawienia się zabarwienia stwarza wrażenie, że osiągnięto punkt końcowy miareczkowania. W rzeczywistości powstanie ciemnego zabarwienia nie jest punktem końcowym, ale odpowiada zaledwie początkowi reakcji, opóźnionemu tylko w czasie z powodu nietypowej kinetyki reakcji Landolta. Punktu końcowego (pierwszego!) w ogóle nie da się w takim miareczkowaniu zaobserwować; odpowiadałby on bowiem momentowi maksymalnej ilości jodu w roztworze (v2). Dalsze miareczkowanie powoduje stopniową konsumpcję jodu przez siarczyn. W momencie drugiego punktu równoważnikowego roztwór odbarwia się. Ponieważ w tym etapie reakcja Landolta przebiega już z dużą szybkością, tempo miareczkowania nie ma wpływu na wynik.
Jak wyjaśnić, że objętość odpowiadająca drugiemu punktowi równoważnikowemu równa jest 1 1/6 objętości pierwszego punktu równoważnikowego v2?
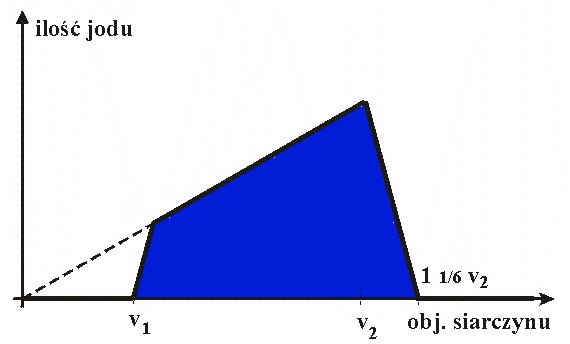
Miareczkowanie to warto wykonać z powodów praktycznych; w ten sposób oszacować można właściwe proporcje substratów dla różnych wersji reakcji Landolta. Przez miareczkowanie roztworu po zakończeniu reakcji, za pomocą roztworu NaOH lub HgCl2 , wyznacza się również maksymalne ilości tych roztworów, które można użyć podczas odpowiednich wersji pokazów.
Dlaczego wynik odwrotnego miareczkowania (w zlewce roztwór siarczynu, w biurecie jodan), nie zależy od tempa miareczkowania? Jak wygłądałyby (dla takiego właśnie miareczkowania) wykres przedstawiający zależność: ilość wolnego jodu w roztworze, w zależności od objętości dodanego jodanu z biurety?
równanie sumaryczne klasycznej wersji reakcji Landolta
W grę wchodzą dwa równania:
2 IO3–
+ 5 HSO3– + H2O → I2 + 5 SO42–
+ 7 H+
2 IO3–
+ 6 HSO3– → 2 I– + 6 SO42–
+ 6 H+ ...(1)
Na pierwszy rzut oka wydaje się, że poprawne jest równanie górne (przecież w reakcji Landolta powstaje wolny jod...). Problem może rozstrzygnąć miareczkowanie. Tym razem posłużyć się jednak trzeba roztworami o dokładnie znanych stężeniach (kłopotliwe jest mianowanie wodorosiarczynu; można wykonać to jodometrycznie). Wynik miareczkowania wskazuje, że substancje reagują dokładnie w proporcji: 1 mol jodanu + 3 mole siarczynu. Tak więc poprawnym równaniem jest dolne równanie (1)! Nie powinno to być zaskoczeniem; do wytworzenia ciemnego zabarwienia trzeba użyć jodanu w ilości wynikającej z równania (1), plus chociażby jedna kropla nadmiaru jodanu. Z powodów praktycznych, w serii wyżej opisanych pokazów używa się jednak znacznie większych ilości jodanu; przebieg reakcji zbliża się więc nieco do stechiometriii opisanej przez równanie górne. W punkcie końcowym powstają znaczne ilości jodu, które nadają mieszaninie ciemne, prawie czarne zabarwienie. Jeśli chce się otrzymać typową dla kompleksu skrobi z jodem błękitną barwę, należy stosować zaledwie niewielki nadmiar jodanu, zbliżony do stechiometrii wynikającej z równania (1). Dla ewentualnego przyspieszenia, można roztwór dodatkowo zakwasić.
"złoty zegarek Landolta" (wersja z arseninami)
Przygotować roztwory:
3 g arseninu NaAsO2 (silna trucizna!) rozpuścić w niewielkiej ilości wody, dodać 16,5 ml kwasu octowego i dopełnić wodą do 150 ml
30 g Na2S2O3 x 5 H2O w 150 ml wody
Zmieszać oba roztwory; po ok. 30 sekundach powstaje złocista zawiesina. Mechanizm reakcji jest złożony i niezbyt dobrze opisany. Prawdopodobnie zachodzi redukcja i dysproporcjonowanie tiosiarczanu do anionów wodorosiarczkowych. Strąca się mieszanina siarczków arsenu oraz siarki.


Ta wersja reakcji zegarowej w rzeczywistości ma bardzo niewiele wspólnego z reakcją Landolta (bez udziału jodanu), jednak omawiana jest czasem w tym właśnie miejscu.
"brunatny pierścionek Landolta"; wędrująca granica
Dotyczy ona rzadko opisywanej wersji, z arseninami w miejsce siarczynu - znanej jako reakcja Roebucka:
5 I– + IO3– + 3 H3AsO3 → 6 I– + 3 H3AsO4
Jeśli reakcję prowadzić w wąskiej probówce (bez mieszania, w obecności skrobi), to wynik zależy od początkowego stosunku ilości reagentów. Przy nadmiarze jodanu, powstaje brunatna strefa utlenienia, wędrująca powoli od górnej powierzchni, w kierunku dna. Przy nadmiarze arseninu powstaje wąski brązowy pierścień stopniowo przesuwający się do dna probówki. Po szczegóły eksperymentalne i wyjaśnienie tego zjawiska samoorganizacji przestrzennej odsyłam do literatury.
zamienniki substratów
W modyfikacjach Reakcji Landolta zamiast wodorosiarczynu można użyć arseninu lub kwasu malonowego. Łatwiej dostępnym reduktorem jest kwas askorbinowy (witamina C). Dopasowanie odpowiednich proporcji reagentów pozostawiam do dobrania empirycznego (podobna procedura opisana jest: ). W odróżnieniu od wodorosiarczynu, produktem utlenienia nie jest mocny kwas (H2SO4), ale słabo kwasowy produkt utlenienia witaminy C. W typowej Reakcji Landolta autokataliza kwasowa jest jednym z głównych czynników lawinowo przyspieszających tempo przemiany. Zastąpienie wodorosiarczynu kwasem askorbinowym zmniejsza więc wyraźnie raptowność momentu końcowego. Zamiennika dla jodanu nie udało mi się jednak zaproponować.
chloran zamiast jodanu...
Zamiast wodorosiarczynu w reakcji Landolta, można użyć arseninu. A gdyby jodan zastąpić chloranem? Stracimy niestety w ten sposób najbardziej efektowną cechę reakcji Landolta: możliwość obserwacji wydzielenia się jodu. Jednak interesujące może być śledzenie przebiegu autokatalizy kwasowej w tym procesie.
Przygotować dwa roztwory:
4,1 g (33,4 mmola) KClO3 oraz 12,6 g (100 mmoli) bezwodnego Na2SO3 rozpuścić w 60 ml ciepłej wody
1,0 ml stężonego H2SO4 rozpuścić w 40 ml wody
Do roztworu mieszaniny siarczynu i chloranu dodać nieco roztworu błękitu bromofenolowego, a potem bardzo powoli, dokładnie mieszając, rozcieńczony kwas siarkowy. Na ściankach wewnętrznych zlewki nie powinna pozostać nie wymieszana z roztworem kropla kwasu. Niezbędne są okulary ochronne: po omyłkowym zbytnim zakwaszeniu reakcja może mieć gwałtowny przebieg! Dodanie opisanej ilości kwasu odpowiada przekształceniu ok. 1/3 słabo alkalicznego siarczynu - w słabo kwaśny wodorosiarczyn. Ostatecznie odczyn mieszaniny zbliżony jest do obojętnego (pH ok. 5). Mieszanina jest zabarwiona fioletowo i można się łatwo przekonać, że takie zakwaszenie jest niewystarczające do zapoczątkowania utleniania chloranem. Fioletowy roztwór rozlać do dwóch cylindrów miarowych 50 ml. Na powierzchnię roztworu w pierwszym cylindrze wkroplić ostrożnie 5 kropel rozcieńczonego (wyjściowego) roztworu kwasu. Po kilkunastu sekundach widać oznaki zachodzenia reakcji: żółta strefa u góry cylindra stopniowo rozszerza się w dół, a roztwór w górnej części cylindra silnie się rozgrzewa (uwaga: temperatura może wzrosnąć do 90o C). Cylindry przed doświadczeniem można z zewnątrz pokryć matową warstewką parafiny, będzie to swego rodzaju prymitywny termometr...
ClO3–
+
2 SO32– + HSO3–
→ Cl– +
3 SO42– +
H+
lub, jeśli ktoś woli:
ClO3–
+ 3 SO32–
+ H+
→ Cl– +
3 SO42– +
H+
natomiast zapis w
formie:
ClO3–
+ 3 SO32– → Cl– +
3 SO42– nie oddaje istotnej właściwości
tego procesu (konieczne jest środowisko kwasowe).
Dodany w górnej części cylindra kwas zostaje co prawda zużyty w reakcji redoks, ale po jej zajściu zostaje wytworzona taka sama ilość jonów wodorowych, które zakwaszają kolejną, przylegającą od dołu warstwę roztworu. W ten sposób dodatek bardzo małej ilości kwasu wystarcza do przemiany całej ilości substratów. A więc jest to typowy przykład autokatalizy. (Powstawanie jonów chlorkowych można wykazać, jeśli do mieszaniny dodać przed reakcją nieco AgNO3)
Do drugiego cylindra dodać również 5 kropel rozcieńczonego roztworu kwasu siarkowego, mieszając jednak natychmiast dokładnie zawartość cylindra. Tym razem reakcja nie zachodzi! Czytelnikowi pozostawiam próbę wytłumaczenia paradoksu: dlaczego w roztworze nie mieszanym reakcja zachodzi, a w roztworze dokładnie wymieszanym - nie?
Opisywane doświadczenie może być przyczynkiem do interesujących rozważań na temat zależności pomiędzy zapisem równania reakcji a rzeczywistym przebiegiem reakcji chemicznej. Dokładniejszy komentarz można znaleźć w pierwszej pozycji literaturowej.
Autokatalizę w utlenianiu chloranami można zresztą zademonstrować prościej: na kawałku blachy wymieszać łyżeczkę sproszkowanego KClO3 z taką samą ilością stałego NaHSO3, dodać kilka kropel wody. Po kilkunastu sekundach rozpoczyna się coraz gwałtowniejsza reakcja, której towarzyszy wydzielanie się dużych ilości silnie drażniącego SO2 (reakcję prowadzić na świeżym powietrzu).
Kinetyka chemiczna uchodzi za jeden z najnudniejszych działów chemii fizycznej... Ciekawe, czy Czytelnik podziela jeszcze ten pogląd?
.
LITERATURA
T.Pluciński.
"Doświadczenia chemiczne" . Wyd. “Adamantan”, Warszawa 1997, s. 60. [Różne
wersje reakcji Landolta, inne reakcje zegarowe]; s. 194.
[Autokataliza w reakcji z chloranami]
Berichte Deutsch. Chem. Ges., 19,
1317 (1886). [Oryginalna praca Landolta]
J.Chem.Educ., 60, 141 (1983).
[Reakcja zegarowa: nadjodan + tiosiarczan]
J.Chem.Educ., 44, A341 (1967).
[Wskaźniki pH w reakcji Landolta]
J.Chem.Educ., 57, 411 (1980).
[Reakcja Harcourta]
J.Chem.Educ., 75, 87 (1998).
[Mechanizm reakcji Harcourta]
J.Chem.Educ., 54, 167 (1977);
69, 236 (1992). ["Old Nassau"]
J.Chem.Educ., 57, 152 (1980).
[Reakcja Landolta z muzyką]
J.Chem.Educ., 61, 1037 (1984).
[Szczegóły mechanizmu wersji "Old Nassau"]
J.Chem.Educ., 76, 530 (1999).
[Wyznaczanie rzędowości reakcji Landolta]
J.Chem.Educ., 60,
494(1983). [Wędrujący front w reakcji jodanu z arseninem]
http://genchem.chem.wisc.edu/demonstrations/Gen_Chem_Pages/12kineticpage/old_nassau.htm ["Old Nassau"]
http://www.mun.ca/educ/ed4361/virtual_academy/campus_a/woodlandm/Demo1.html [j.w.]
http://www.chem.leeds.ac.uk/delights/texts/Demonstration_10.htm [Reakcja Landolta]
http://chem.lepeer.org.Chem2Docs/Rate.Eq.html [Kinetyka reakcji nadsiarczanowej]
L (strona
nieczynna)
http://chemlearn.chem.indiana.edu/demos/Hydrogen.htm [Reakcja Harcourta]
Tomasz Pluciński
nowy adres:
tomasz.plucinski@ug.edu.pl
| F | strona główna |
{kind=link}